
Neuromuscular Disorder As A Chronic Disease And Its Impact On Quality Of Life


Abstract
Numerous diseases are categorized as chronic disorders. These can be related to neurological disorders, movement disorders, respiratory diseases, cancer, autoimmune disorders, and genetic disorders. Chronic disorders are a prime contribution in deaths occurring below 70 years of age. Functional disability scale was use to measure the hardships associated with disease that indirectly affect the routine life of the sufferers. Charcot-Marie-Tooth disease type 1A (CMT1A) is the most common subtype of inherited peripheral neuropathy characterized by distal muscle weakness, atrophy and sensory loss. CMT1A is caused by the duplication of PMP22 gene on chromosome 17p12 region. However, despite this evident cause, onset age or severity is considerably variable among patients. The exact reason behind these phenotypic heterogeneities is rarely discovered yet, but certain secondary factors are assumed to be involved. Since miRNAs are the key regulators of gene expression, we speculated variants of miRNAs might be the genetic modifiers for CMT1A. This study noticed a common single nucleotide polymorphism (SNP) of miR-149(rs2292832) as a possible candidate. MiR-149 was predicted to target several CMT causing genes including SH3TC2, LITAF, MPZ, and PMP22 by several prediction algorisms. The rs2292832 was located near the 3' end of precursor, the important region for maturation. From the association study for 176 unrelated Korean CMT1A patients, we identified that the TC/CC genotypes of rs2292832 were significantly associated with late onset (onset age ≥20) and mild phenotype (functional disability scale 0-2, CMT neuropathy score 0-10). Therefore, this study suggests that themiR-149SNP has the considerable potential to affect the phenotypic heterogeneity of CMT1A as genetic modifiers.
Keywords: Chronic diseaseCMT diseaseperipheral myelin protein 22functional disability scale
Introduction
A disease that lasts three or more than three months and ends up with death is known as chronic disorder. In other words, it is long lasting and symptoms of the disease become vigorous with the passage of time. Some of the most familiar conditions are asthma, diabetes, epilepsy, HIV/AIDS, hepatitis, some genetic disorders, chronic obstructive pulmonary disease, brain associated disorders, hearing loss or deafness, arthritis, or it can be heart related, or cancer,
On the basis of the biomedical classification, chronic disorders can be defined as the disease that spans more than three months or more according to the U.S. National Institute of Health (NIH, 2016). Chronic disorders cannot be treated by medication nor do they disappear. However, symptomatic treatment can be used (Bauer, Briss, Goodman, & Bowman, 2014). Numerous diseases are categorized as chronic disorders and these can be related to neurological disorders, movement disorders, respiratory diseases, cancer, autoimmune disorders, genetic disorders and so on. However, this is not the complete list (Abegunde, Mathers, Adam, Ortegon, & Strong, 2007). Aging itself is a disease from a certain point of view. In developed countries, aging of populations is getting pronounced as these countries have a higher life expectancy. As a result, they have a higher aged population (Manton, 1988). A higher aged population will result in more chronic patients which, in turn, would need more funds are required for prolonged treatment to enjoy the quality of life like normal people. (Lim et al., 2007). Almost 25 % of the population of the United States of America suffer chronic disorders and need prolonged treatment, rehabilitation and some siding material that requires more funds (Ward & Black, 2016).
Chronic disorders are the main cause in a person’s life that can directly affect the settled routine of life that is enjoyed without the disorders. The susceptibility of a chronic disease gets higher with the aging as the majority of the diseases only appear in old age. However, it is not always limited to old age. Some of the acquired or environmental factors also contribute for such illness as well. Chronic illness is also known as non-communicable diseases according to the World Health Organization (WHO).
Neuromuscular Disorders
Neuromuscular disorders affect PNS and muscular activity. The foremost effect of Charoct-Marie-Tooth disease is that it disturbs the voluntary movement. It is a progressive disorder and the affected extremities are primarily those controlled by the sensory and motor neurons (Morrison, 2016). Some of the neuromuscular diseases are associated with CNS; however, most of the them are related to PNS. Due to lack of coordination between nerve cells, the individual’s physical activity becomes limited which directly impacts on quality of life. Due to muscular dystrophy caused by genetic causes, muscular mass is reduced which comes under the specific category of muscular turmoil known as Charcot_Marie-Tooth disease (CMT) (McDonald, 2002). CMT refers to a cluster of innate disorders of peripheral neuropathies where motor or sensory nerve cells are damaged. CMT disease is primarily classified into two categories called CMT1 (Demyelination form) and CMT2 (axonal form).
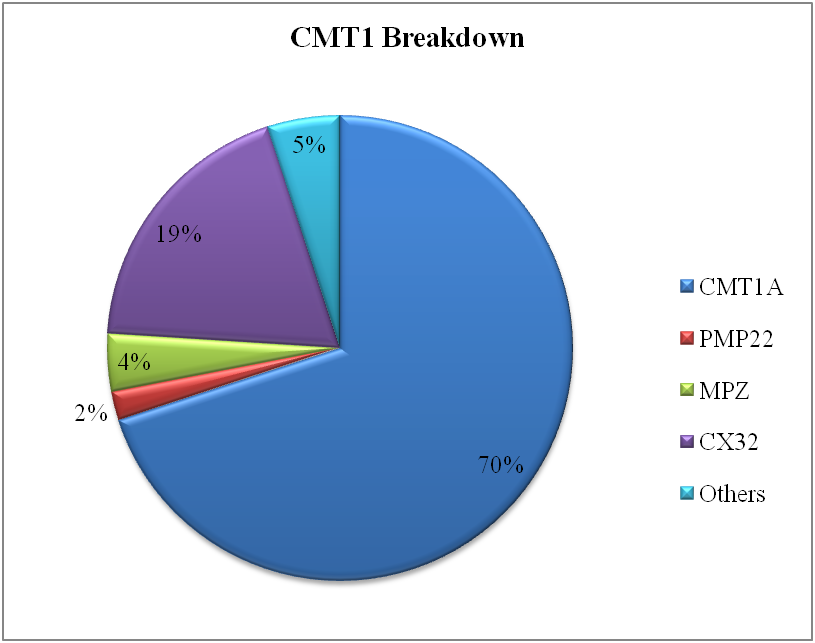
Charcot-Marie-Tooth disease is most widespread hereditarily multiple gene disease with the frequency between 4.7-36/100,000 (Krajewski et al., 2000). As the motor neurons are affected, so the predominantly phenotype is distal muscle weakness of both upper and lower limbs. Sometimes, this is also called muscular dystrophy (Vallat & Funalot, 2010). As the disorder is heterogeneous, almost 80 genes are reported to be associated with the CMT. Several genes responsible for the disease made protein that have a key role in trafficking involved between the cells. The genes involved in interacellular tracking are the most notorious for causing the disorder (Birouk, 2014).
Charcot-Marie-Tooth disease (CMT) is distinguished by the failure and atrophy of the large extremity muscles, and sensory loss (Dyck & Lambert, 1968) The disease was identified by two physicians named Charoct and Marie in Paris (1886) and a third scientist called Tooth individually identified the same disease in London (UK) in the same year (Pareyson, Scaioli, & Laura, 2006). Categorization is bottomed on a mixture of neurophysiologic distinctiveness, legacy model, and fundamental inherited cause. Clinical findings based on nerve shape and NCV and on three diverse subtypes are well-known, i.e. the demyelinating type (CMT1) can be sorted out on the basis of the speed of the nerve traveling down the nerve cells that is below 38 m/s (see figure
CMT type 1A
CMT type 1A is rooted by replica of 1.5mb, or alteration in, the gene encoding peripheral myelin protein22. On the whole, occurrence of CMT is typically reported as 1:2,500. However most of the disease is caused by the pmp22 duplication. Onset age of CMT1A is the initial two decades of life with difficulty walking or running. Extremities are usually most affected.
Problem Statement
According to the literature, in various stages of the life, it has been found that although type 1A is the outcome of the duplication in peripheral myelin protein 22 gene, the severity of the disease has a wide range from almost normal to bed ridden. As all patients were found to have the same reason for the origin of the disease, they however demonstrated variations in symptoms in terms of onset age according to the Functional disability scale (FDS). To find out the exact principle behind this ambiguity, we hypothesized that there may be some genetic modifier or some microRNAs involved that can alter the expression of the protein. FDS was applied to each patient according to the following criteria: 0, normal; 1, normal but with cramps and fatigability; 2, unable to run; 3, difficulty walking but still able to walk unaided; 4, walking with a cane; 5, walking with crutches; 6, walking with a walking frame; 7, wheelchair bound; and 8, bedridden.
Research Questions
Charcot-Marie-Tooth disease type 1A (CMT1A) is one of the most common inherited neurological disorders, evidently caused by duplication of a 1.4 Mb region on chromosome 17p12, so that, theoretically, 1.5 increased dosage of the peripheral myelin protein22 (
We noticed microRNA (miRNA), one of the key regulators of gene expression, as a candidate of modifier.miRNAs are a class of small non-coding RNAs that regulate the expression of multiple target mRNAs by binding to the 3' untranslated region (3'UTR) of targets and inducing their degradation or translational inhibition(Bartel, 2009).Various studies have identified miRNAs are critically involved in various biological and pathological processes including cell proliferation, differentiation, apoptosis, and pathogenesis of several diseases (Sayed & Abdellatif, 2011). Since SNPs located in pre-miR genes may affect the miRNA maturation, expression or the binding to target mRNAs, and subsequently alter the expression of target genes (Jin & Lee, 2013), we speculated SNPs on miRNAs that can regulate PMP22 expression directly or indirectly also have the potential to control the CMT1A phenotypes.
In the current study, we hypothesised that miRNAs that can play a critical role by targeting the PMP22 or other CMT related genes and that the expression of the miRNAs can affect the expression of the protein which can then alter the end result of the gene in terms of phenotype or onset age. MiR-149 has been found to be associated to various CMT genes as a genetic modifier and a SNP rs2292832 close to the 3’ end can also alter the expression.
It has been found that it also act as a regulator for many genes and diseases (Bischoff et al., 2014; Wang et al., 2014). So, we selected miR-149 as a candidate to demonstrate the association between its SNP and diverse CMT1A phenotypes.
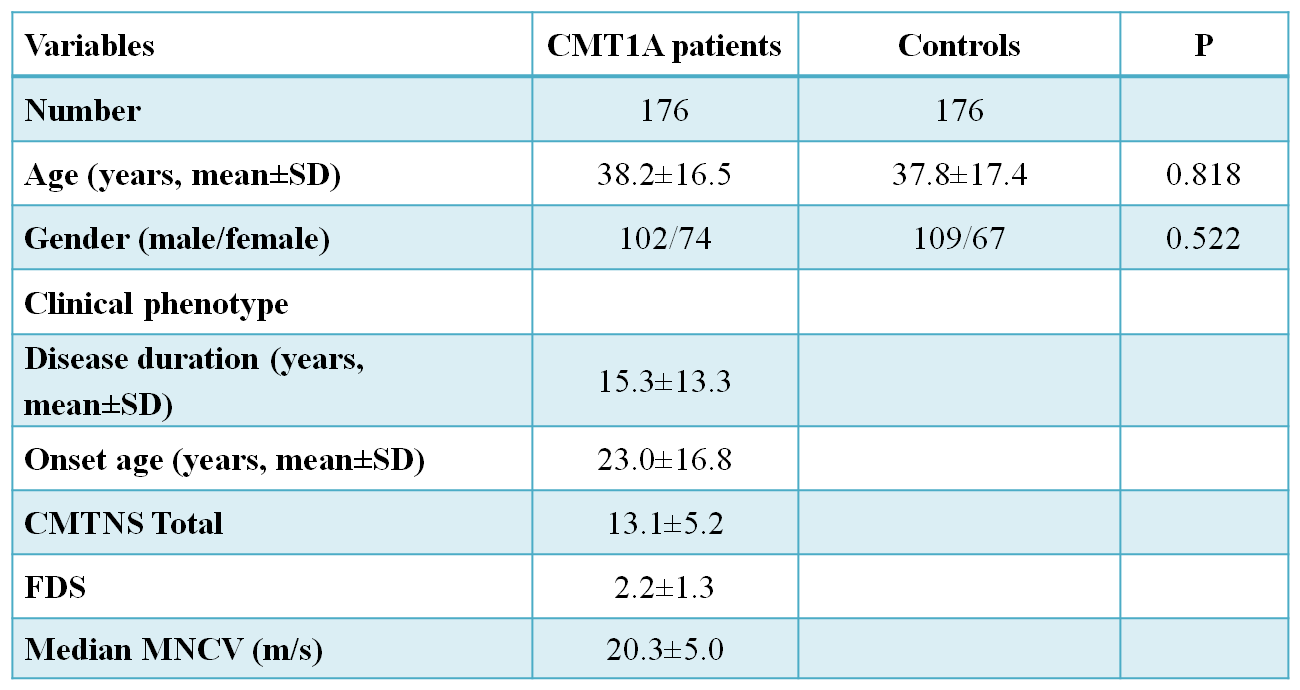
Base-line characteristics of CMT1A patients and control subjects are presented in Table
Purpose of the Study
CMT disease is one of the most prevalent peripheral disorders that can affect the life of the individual as well as the family and community. To identify the underlying cause of the severity of the disease and its impact on quality of life was the main focus of this study. The quality of the life was determined by the Functional Disability Scale. FDS was applied to each patient using the following criteria: 0, normal; 1, normal but with cramps and fatigability; 2, unable to run; 3, difficulty walking but still able to walk unaided; 4, walking with a cane; 5, walking with crutches; 6, walking with a walking frame; 7, wheelchair bound; and 8, bedridden (Boentert et al., 2014)
Research Methods
This study included 176 Korean unrelated CMT1A patients and 176 healthy controls that had no clinical features of neuromuscular disorderTotal DNA was extracted from whole blood using QIAampDNA blood kits (Qiagen, Hilden, Germany). The CMT1Aduplication was determined by genotyping six microsatellites(D17S921, D17S9B, D17S9A, D17S918, D17S4A,and D17S2230) within the 1.4 Mb duplication region in chromosome 17p12 by hexaplex PCR (Choi et al., 2007). Duplication of
The current study incorporated several unrelated CMT1A patients and healthy samples without any symptoms of neuromuscular disorder were selected as the control sample for comparisons between the affected and control samples (figure
Whole blood samples were taken from the patients and control sample by using the QIAampDNA blood kits (Qiagen, Hilden, Germany). The first step after collecting the DNA was the determination of the CMT1A duplication using the six microsatellite markers in the 1.4Mb region of the chromosome 17p12 using the hexaplex PCR. Duplication was determined by real-time quantitative PCR. All the participants were given written informed consent according to the standard protocol approved by the IBR for Sungkyunkwan University, Samsung Medical Center.
Findings
From various target prediction algorisms, miR-149 was estimated to mark several CMT-causing genes including PMP22, SH3TC2, MPZ and LITAF. A common SNP, rs2292832 (NR_029702.1:n.86T>C) was discovered near the 3'end of precursor from our 356 Korean samples WES and public databases such as 1000 genomes and dbSNP. The 1000 genomes minor allele frequency of rs2292832was 0.613. The predicted secondary structure and the scheme of interaction of miR-149with some target genes are presented in Figure
To investigate the relevance of rs2292832 and disease onset age, we stratified CMT1A patients into two subgroups depending on the onset age. The genotype occurrence of rs2292832 in early onset group (<20) deviated remarkably from HWE (P=0.036) (Figure
To investigate the association between miR-149 genotype and onset age properly, we compared the genotype frequencies of two clusters. The data showed an obvious relationship between the two SNPs and onset age. Under the dominant model, the frequencies of TC/CC genotypes in the late onset group (≥20) were significantly higher than in the early onset group (<20) (TT versus TC+CC). From this data, we could deduce that the frequencies of C allele of rs2292832 were higher in the late onset group.
Our focus was to identify to what extent the quality of life of a patient is disturbed when suffering from a neuromuscular disorder. The Functional disability scale was used to measure the hardships associated with the disease that indirectly affects the routine life of the sufferers. It was found that almost 70% of the patients have 3-7 functional disability score that is far beyond the normal value. With FDS score of even 3 or above, it becomes very difficult to walk easily as either you need crutches or some walking aid.
Conclusions
The frequency of NCDs have a devastating impact on health which ultimately affects the life of individual, caregivers and community as well. The major risk factors for the NCDs are smoking, alcohol, physical sluggishness and unhealthy diet. We can control NCDs by avoiding the risk factors mentioned above. A healthy lifestyle plays a major role to avoid NCDs even in advanced age. However, there are some NCDs which result in health deterioration caused by genetic factors. Genetic disorders can also be controlled by genetic screening. In this way, we may be able to save our next generation form inherited chronic disorders like CMT.
Our findings show that C alleles of the rs2292832 are strongly linked with late onset and late onset was associated with milder symptoms of the disease. To conclude, our findings show that C allele were strongly associated to late onset, and also markedly related to mild phenotypes in the case of late onset. As explained earlier, some reports have mentioned that alterations in rs2292832 may modify the expression level of the established structure. So we can assume that there is a possibility that the mutations of the single nucleotide polymorphism locus can result in the decrease of the mir-149 appearance, pursued by an increase of target genes.
Current data of associated studies of SNP in miRNA constantly indicated that affected persons with miR-149 alterations had an affinity of moderate symptoms of the disease. Hence, the current study and previous reports about miR-149 concur. In other words miR-149 can alter the disease (CMT1A) symptoms without affecting the expression of the PMP22 gene.
However, our associated study data consistently indicated that the patients with miR-149 mutations had a tendency of milder phenotype. Thus, according to preceding information and our data as they stand, miR-149 may affect CMT1A symptoms not by direct targeting of PMP22 gene but, in an indirect way, however, it can affect the expression of the protein by targeting the other factors involved in the expression of the PMP22 protein.
As reported in the literature, miR-149 has the possibility to be involved in skeletal muscle disorders, hence, it is probable that alteration in the miR-149 may be responsible for the CMT1A phenotype by the route of muscle metabolism.
In wrapping up, we find evident correlations flanked by the genotypes of miR-149 rs2292832 and the phenotypes of CMT1A from this study, although the direct relationship of miR-149 and PMP22 was not observed. This indicates vast possibilities that miR-149 could affect the relative seriousness of CMT1A by regulating certain mechanisms of the peripheral nerve system. Thus, the verification the definite target of miR-149 and their clear functions by further experiments is imperative.
References
- Abegunde, D. O., Mathers, C. D., Adam, T., Ortegon, M., & Strong, K. (2007). The burden and costs of chronic diseases in low-income and middle-income countries. Lancet, 370(9603), 1929-1938. Retrieved from doi:10.1016/s0140-6736(07)61696-1
- Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell, 136(2), 215-233. Retrieved from doi:10.1016/j.cell.2009.01.002
- Bauer, U. E., Briss, P. A., Goodman, R. A., & Bowman, B. A. (2014). Prevention of chronic disease in the 21st century: elimination of the leading preventable causes of premature death and disability in the USA. Lancet, 384(9937), 45-52. Retrieved from doi:10.1016/s0140-6736(14)60648-6
- Birouk, N. (2014). [Review of the recent literature on hereditary neuropathies]. Rev Neurol (Paris), 170(12), 846-849. Retrieved from doi:10.1016/j.neurol.2014.10.001
- Bischoff, A., Huck, B., Keller, B., Strotbek, M., Schmid, S., Boerries, M., . . . Olayioye, M. A. (2014). miR149 functions as a tumor suppressor by controlling breast epithelial cell migration and invasion. Cancer Res, 74(18), 5256-5265. Retrieved from doi:10.1158/0008-5472.can-13-3319
- Boentert, M., Knop, K., Schuhmacher, C., Gess, B., Okegwo, A., & Young, P. (2014). Sleep disorders in Charcot-Marie-Tooth disease type 1. J Neurol Neurosurg Psychiatry, 85(3), 319-325. Retrieved from doi:10.1136/jnnp-2013-305296
- Brewer, M. H., Ma, K. H., Beecham, G. W., Gopinath, C., Baas, F., Choi, B. O., . . . Antonellis, A. (2014). Haplotype-specific modulation of a SOX10/CREB response element at the Charcot-Marie-Tooth disease type 4C locus SH3TC2. Hum Mol Genet, 23(19), 5171-5187. Retrieved from doi:10.1093/hmg/ddu240
- Choi, B. O., Kim, J., Lee, K. L., Yu, J. S., Hwang, J. H., & Chung, K. W. (2007). Rapid diagnosis of CMT1A duplications and HNPP deletions by multiplex microsatellite PCR. Mol Cells, 23(1), 39-48.
- Dyck, P. J., & Lambert, E. H. (1968). Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol, 18(6), 603-618.
- Harding, A. E., & Thomas, P. K. (1980). The clinical features of hereditary motor and sensory neuropathy types I and II. Brain, 103(2), 259-280.
- Jin, Y., & Lee, C. G. (2013). Single Nucleotide Polymorphisms Associated with MicroRNA Regulation. Biomolecules, 3(2), 287-302. Retrieved from doi:10.3390/biom3020287
- Kiyosawa, H., Lensch, M. W., & Chance, P. F. (1995). Analysis of the CMT1A-REP repeat: mapping crossover breakpoints in CMT1A and HNPP. Hum Mol Genet, 4(12), 2327-2334.
- Krajewski, K. M., Lewis, R. A., Fuerst, D. R., Turansky, C., Hinderer, S. R., Garbern, J., . . . Shy, M. E. (2000). Neurological dysfunction and axonal degeneration in Charcot–Marie–Tooth disease type 1A. Brain, 123(7), 1516-1527. Retrieved from doi:10.1093/brain/123.7.1516
- Lim, S. S., Gaziano, T. A., Gakidou, E., Reddy, K. S., Farzadfar, F., Lozano, R., & Rodgers, A. (2007). Prevention of cardiovascular disease in high-risk individuals in low-income and middle-income countries: health effects and costs. Lancet, 370(9604), 2054-2062. Retrieved from doi:10.1016/s0140-6736(07)61699-7
- Lupski, J. R., de Oca-Luna, R. M., Slaugenhaupt, S., Pentao, L., Guzzetta, V., Trask, B. J., . . . Patel, P. I. (1991). DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell, 66(2), 219-232.
- Manton, K. G. (1988). The global impact of noncommunicable diseases: estimates and projections. World Health Stat Q, 41(3-4), 255-266.
- McDonald, C. M. (2002). Physical activity, health impairments, and disability in neuromuscular disease. Am J Phys Med Rehabil, 81(11 Suppl), S108-120. Retrieved from doi:10.1097/01.phm.0000029767.43578.3c
- Morrison, B. M. (2016). Neuromuscular Diseases. Semin Neurol, 36(5), 409-418. Retrieved from doi:10.1055/s-0036-1586263
- Pareyson, D., Scaioli, V., & Laura, M. (2006). Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromolecular Med, 8(1-2), 3-22. Retrieved from doi:10.1385/nmm:8:1:123
- Sayed, D., & Abdellatif, M. (2011). MicroRNAs in development and disease. Physiol Rev, 91(3), 827-887. Retrieved from doi:10.1152/physrev.00006.2010
- Sinkiewicz-Darol, E., Lacerda, A. F., Kostera-Pruszczyk, A., Potulska-Chromik, A., Sokolowska, B., Kabzinska, D., . . . Kochanski, A. (2015). The LITAF/SIMPLE I92V sequence variant results in an earlier age of onset of CMT1A/HNPP diseases. Neurogenetics, 16(1), 27-32. Retrieved from doi:10.1007/s10048-014-0426-9
- Vallat, J. M., & Funalot, B. (2010). [Charcot-Marie-Tooth (CMT) disease: an update]. Med Sci (Paris), 26(10), 842-847. Retrieved from doi:10.1051/medsci/20102610842
- Wang, Z., Wei, M., Ren, Y., Liu, H., Wang, M., Shi, K., & Jiang, H. (2014). miR149 rs71428439 polymorphism and risk of clear cell renal cell carcinoma: a case-control study. Tumour Biol, 35(12), 12127-12130. Retrieved from doi:10.1007/s13277-014-2517-5
- Ward, B. W., & Black, L. I. (2016). State and Regional Prevalence of Diagnosed Multiple Chronic Conditions Among Adults Aged >/=18 Years - United States, 2014. MMWR Morb Mortal Wkly Rep, 65(29), 735-738. Retrieved from doi:10.15585/mmwr.mm6529a3
Copyright information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
About this article
Publication Date
29 October 2018
Article Doi
eBook ISBN
978-1-80296-046-4
Publisher
Future Academy
Volume
47
Print ISBN (optional)
-
Edition Number
1st Edition
Pages
1-75
Subjects
Psychology, clinical psychology, psychotherapy, abnormal psychology
Cite this article as:
Kanwal, S., & Perveen, S. (2018). Neuromuscular Disorder As A Chronic Disease And Its Impact On Quality Of Life. In Z. Bekirogullari, M. Y. Minas, R. X. Thambusamy, & C. Albuquerque (Eds.), Clinical and Counselling Psychology - CPSYC 2018, vol 47. European Proceedings of Social and Behavioural Sciences (pp. 67-75). Future Academy. https://doi.org/10.15405/epsbs.2018.10.6